ncbi更新后怎么搜索基因组数据
给大家来介绍一下,嗯,怎么查找这个物种的一个金组有没有发表?我们今天用的是 n c b i 的数据库,然后在这里这个 这个就是我们的这个金组的情况。我们以人类为例,搜索一下之后呢,他会出来, 因为 n c n c b i 数据库的话,基本上都是加载比较慢的情况,所以大家如果要用的话可以,嗯,就是不要晚上用,然后 大致在白天或者说是,嗯,就科研工作者时间少的情况下,嗯,然后这个还会好一些。 这个呢是我通过刚刚之前没有搜索库,然后来去获得的。像如果说没有库的话,他会直接就告诉你整个没有设定数据库的话,整个他的这个数据都会出来, 像基因的蛋白的这个就是我们的基因组的一个情况,还有你看他一些临床的数据和 pop, 嗯, pop chemistry 的数据也会放到这里面。 另外呢我们还可以通过首页,然后点 这里直接进入这个金锁的这个数据库,然后点这里可以直接搜,直接看那个我们的这个物种,或者说是直接按照物种来查, 他这个是表格的一个形式,就是跟之这个的话,就是有点像之前 k e g g 的那个物种的一个情况,是以表格的方式来列出的, 他这个在加载,然后我们可以通过这里去筛选这个按钮,比如说我们选一个 好 细菌了吧, 啊,这样这里就可以看到看他的这个细菌,他的一个组织的或者说物质物种的名字,然后他的一个分类情况。 另外就是他的这个金组的大小以及染色体的多少,这种你看这个细菌的话,他基本就是一的这种情况。另外这个他有没有纸粒啊?等等。那我们选一个 真菌,或者说是这个 其他的这种他就会有一些。你看他的这种针和生物的话,他有的染色体就是多种类的,这个应该是一种鱼吗?鱼类的,然后他的染色体条数的话就是二十五 这种情况。然后直,然后比如说你想要那个的话,直接点这个 恰恰, 或者说直接点进去看刚下载的内容,直接 这个应该就是这一页的。看这个,是啊,这个是整个下下来的,就是我们整个搜索到的一个结果,一万一万两千多, 这个的话一页是五十,然后乘二百四十五,就大概一万两千多,就这个分类里面的整个他的都是下下来的 啊,这种单独的话就是可以点击,然后下这个话。

粉丝99获赞527
相关视频
 00:20查看AI文稿AI文稿
00:20查看AI文稿AI文稿输入需要查阅的目的基因,点击回车,右侧显示的是部分功能区,中间为该基因的简单信息,点击后右侧列出整个网页的目录, 右侧为该基因详细信息、位置、图谱表达量、相关研究文献、功能注视等。嘎嘎好用。
468译帆英文论文润色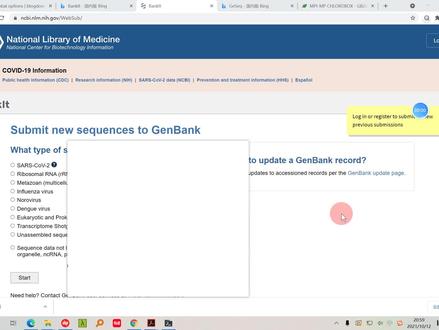 12:27查看AI文稿AI文稿
12:27查看AI文稿AI文稿大家好,我是小明,今天来录制期视频,跟大家简单介绍一下如何向 ncbi 提交叶立体或者线立体的金组训练啊。我们 再把样品测完序,然后经过组装,首先我们会拿到一个完整的液力体进度的序列,发磁 a 格式或者线立体进度的发磁 a 格式啊,就是这个样子的,然后 拿到发磁器格式以后,我们会对他进行注视,注视以后通常会拿到一个近半个格式的文件,就是这个 啊,现在我们手头如果有了这两条这两个序列文件的话,然后接下来就是把 gba 文件转化成 tbl 格式的文件,然后使用这个 b a klt 这个工具,一个在线工具,然后向 ncb i 提交 啊,现在已经假设我们已经拿到了发子这一盒进行半个格式的文件,然后接下来这一步就是如何准备 tbl 格式的文件。 tvl 大概给大家看一下他那个文,基本的文件格式就是这个样子的, 首先第一行是大约号开头一个 c 顺,这个是固定的,然后后面跟一个训练名字,然后后面就是跟你那个训练侧轴,比如这个就是一个 大档口,回去一到什么什么,然后这个是一个基因,然后基因的名字,然后 cds, 然后产物什么基的名字,这样依次排列下来的。这个 这个文件是我们可以直接通过这个机币文件得到的,然后我们可以借助这个在线工具,就是这个 gecco, 我们直接在这个 b 应搜索引擎里面搜 gecco, 然后第一条就是这个工具,我们点开以后就是这样子的,我们直接 使用 这个,这个叫 好,我点一下他应该打开以后,他是 这个注视界面,然后我们选这个 gb c 可 uni 这个的,我们点这个,然后他会有一个转换的界面,然后这个地方我们就直接选选择我们的 gb 文件, 然后接下来就是 选这个,因为业立体或者先立体进度通常都是缓装的,然后这个一张密码进行,我们也先不用改啊,这个地方会选业立体或者先立体,这个地方自己选一下,然后直接点这个开始转换就行了。 这个通常很快,我们稍微等一会,然后这个台位有两个报,就是有一些报错或者警告信息,这个第一个错误就是其实明码子错误,但是这个就可以把他忽略到了,因为有的接他可能不是以那个 adg 开头的, 其实所以他就会有这个内容啊,这个就是下面这个也是错的蛋白,其实这两个应该是一个意思吧。 好,下面这警告信息就不用管了。呃,然后但是如果你这个错误的信息特别多的话,你后续提交可能会有错误,这个大家需要注视,需要注意一下,就是自己那个经营办法格式的文件 里面的注视内容需要是没有错误的后才行才能提交。然后我们需要用到的就是这个我能提神提宝,然后把这个点击, 然后就把这个下载下来就可以了。现在这个就是 tbl 格式的文件,这个提交的时候需要注意点,就是 取消的时候我们需要用到 facec 格式,然后还有这个 tbl 格式,然后这个 gpv 文件我们就后续就不需要了,他这个需要把这个表头对准给他改一下,他这表头的基本内容就是第一行是一,第一个是一个训练名,然后这个空格中框里面是固定的,这就是这个 前面是固定的,后面接这个物种的拉丁名,然后这个拉丁名,然后叶类体完整的间组,如果是县令的话,你就把这个单词改成县令就行。然后这个 tbl 格式的文件是 他这个肥,这是固定的,然后这个这个一对应的是发 c 文件的那个序列名就是这个,这个是 什么内容啊?飞制后面也要对应的跟什么内容。现在这两个文件准备好了,接下来我们就是要提交内容, 我们直接在这个冰搜索引擎里面搜这个 b a k 二 t, 就这个,然后第一条跳出来的就是这个在线奇效工具。然后这个的话我们要用的话,首先第一步要登录一下,我记得之前 ncbi 是可以单独注册账号的,但是 最近一次我登录的时候,他提示我就不能不让用之前那个账号了,所以他要用一个第三方的登录,他这个有很多内容,不是下面有这么多内容可以登录, 那我就直接用这个酷酷狗账号来登录了,因为电脑上是登录了这个酷狗账号的,然后直接就点这个 就可以了 啊,这个大家如果没有科学上网攻击的话,可能 有时候会稍微有点慢 啊。现在这个我就已经登录上了。好,登录上的话在这边就有很多可以选,因为我们提交的是细胞基金组的内容,就直接选这个,最后一个 就是这个里面是包癌细胞剂监督内容的,然后点这个丝带的开始就行了 啊。我准备这个百字方块是因为最开始有一些呃个人信息,我所以需要把它盖住,然后我们直接点这个开始提交就可以了。这个是我之前提交的一些内容,把它放到这要把我的个人信息盖住, 因为刚才我试了一下这个不继续就行, 就想刚才这个没有完成的奇效,怎么把它删掉呢? 这个没有完成的提交还只能继续正常你那个值,如果没有这个的话,就直接点这个 start 提交就可以了,然后那我就直接点这个继续就行了。他第一步应该是跳出来这个, 这个挡住底部跳出来的是这个,然后后面就会让你填一些个人信息 在这些这个里面,然后你就按照他这个英文单词是什么意思,然后就填什么内容就可以了。 直接点这个继续,后面应该就不涉及到个人信息了,把这个关掉, 然后这个地方会挑这个填序列的作者,序列的作者,然后这个是说直接填我的名字,就往这上填,然后如果还有其他人的话,你可以往上继续再往上添加就行了,如果没有的话就把这个填掉进了。然后接下来就是 问你这个训练是不是已经发表,然后通常体校训练的时候就是都是没有发表的,然后就选这个就行了,然后会填一个标题啊,这个标题你自己随便写就行了,比如这个列文就叫 完整的 什么什么什么这个名字,然后还有接下来就 是这个参参考文件的作者,这个作者的话你可以选和训练的作者一样,或者单独指定新的作者也行,然后出个单独指定新作者,你选这个,他下回就会有这个框,如果选这个也可以选这个是也可以,然后直接就继续, 然后他就会让你选测序平台,作业立体的通常都是羽绒蜜蜡测序或者后面做线立体。用三代的话,你可以选这个白白白啊,或者这个应该也是三代测序的一个平台吧,然后或者都没有的话,你就选这个二字, 然后这个地方就是直接选这个一路面到了,然后选组装好的序列,然后这个地方会填这个组装程序。 呃,一类游线立体通常比较常用的就是这个这个工具,然后会后面添一个版本,然后就直接点继续就行了, 然后他这个地方会让你选释放日期,然后通常就会选这个处理后直接释放,但是虽然是处理后直接释放的话,他这个也会要求很长时间,你看根据他下面这个,如果你自定义这个释放日期的话,他也会要求你 这个日期必须在提交日期之后的六个月之后或者十年之内。然后接下来就选这个分子类型,这个地方,这是锦囊那个点,然后他的那个结构,这个叶类底部建立童装都是缓装的,我们就直接选这个缓装的 这个问题是否提交了一个完整的七八七基因组?我们选择是, 然后就是提交序列,他这个地方可以选择,你把序列粘过来,或者直接选择文件,这个地方我们就直接 选择文件就行了,选择你的发 c 文件,然后点 ctrl, 他就会上传系列, 然后这个再点一次 ctrl, 这个就直接选这个, 他问你是否是原始题型或者是第三方注视题型,直接选这个二维震动就行了,然后点继续, 然后这个地方会选一个吸霸器的位置,然后就是液类体,或者你就选线类体也行,然后这个 这个 sosos modeper, 这个具体什么意思我还没太搞明白,但是之前做果树的时候,果树通常都会设计一个品种,所以他 这个地方可以选,他选卡好体位,看这前面有一个品种, 但是你如果有什么特殊的的话,他好像可以选,就这个是品种,然后你可以选什么有的物种可能有什么生态味型啊,然后畸形之类的这个东西,然后就在这填就行了,这个东西好像不选应该也行, 这个地方我就给他扣, 我记得上次提奥没上,然后把他刷新一下,这个地方就给他空着就行, 这个把它旋上叶罗丽体, 然后你再看下面这个就是序列的 id, 然后这个是对应的那个拉丁名,这个就是根据你那个发 c 格式文件最开始定义的 那个名字,他自己生成的,我们点击肯定就行了。然后接下来就是添加注视文件,这个我们就是选择第一个就是五列的这个填充表,就是那个点 tbl 格式的那个内容,然后直接选择文件,然后选这个, 然后接下来点这个上传, 上传以后他就会生成这个内容,然后在下面还会有一个 在这个地方有 ctrl, 然后下面是根据你这个上传的 tbl 内容,然后他自动生成的一个 tb 格式的文件, 我们直接点继续。 当然如果你那个 tpl 格式的 文件准备的就是最开始的击背文件,就是有错误的话,刚才那一步可能就会有错误,然后他你就根据他那个提示来改就行了。 然后现在就是最后一步,直接我们就点这个飞那三分没认就可以了,但是因为这个我之前提交过了,所以这个地方就不点了,然后这块添一个邮箱,然后 相当于是提交,整个提交过程就完成了。这个地方我用的是叶立体金组做的介绍啊,线立体金组是同样的步骤,完全是一样的 啊。然后今天的内容就介绍这么多,大家如果觉得视频内容还有帮助的话,可以点赞关注,投币转发进行支持。也欢迎大家关注我的公众号,小明的数据分析笔记本,留言讨论本期视频相关内容,谢谢大家的收看。
 12:28查看AI文稿AI文稿
12:28查看AI文稿AI文稿这节课程我们来介绍一下 ncbi 数据上传,在数据处理完成之后,一般发表文章需要公开数据,因此将数据上传 ncbi 是一项重要的工作。对于 ncbi 数据上传, ncbi 官网都有详细的说明文档, 但是上传的步骤过于繁琐,新用户第一次操作还是需要非常大的学习成本。 ncbi 上传数据最重要的是两点,第一点是填好正确的信息,第二点是数据格式一定要满足 ncbi 的要求。 那么接下来我们就为大家介绍一下如何来进行 ncbi 数据的上传。首先我们来看一下可以上传到 ncbi 的数据类型, 这里面列出了 ncbr 可以接受的数据类型以及支持的上传方法, 包括拼接好的金主完成图草图、转录组数据、高通量测序位置、金主变异数据以及单条测距数据等等类型。这些数据根据不同的数据类型上传到 ncbi 不同的数据库中, 例如金主信息上传到知音、 bugriphone 等数据库,转录组数据上传到 tsa 数据库,而车区锐志则上传到 sra 数据库。下面我们来介绍一下上传数据过程需要使用的工具, 上含的工具包括钢铁的和筛和印。如果是比较小的 数据,一种十六 s 设计数据、单调基因等可以使用半 k 的在线提交就行。这里面注意,刚给的也并不是所有小的数据都可以接受, 如果剪辑小于两百 bp 就不行,除非标明这些数据来自于外弦指,外弦指有可能小于两百 bp 或者 ncra 小 ra 等。 如果是 e s t 数据,需要使用 d b, e s, t, c, c, t, c s s 数据,需要使用 d b, z, s, s, c。 四头 sts 数据需要使用 dbsts, cston 都不能使用班级的系统。 赛克印并不是网页使用,而是安装到本地电脑的软件,所以功能更多,支持的数据类型也更多, 可以完成批量上传。还有一个数据转换的工具, tbl two a s n tbl two a s a 是命令行软件,用于把序列处置信息转化为点 sq 文件,这个文件在上传金主序列的时候是必须用到的。 在数据上传过程中需要填写很多的信息,最好提前把这些信息整理好。包括联系信息,例如上传者的姓名、工作单位、部门、单位地址、日期、电话号码、邮箱等。 序列信息包括物种名、样品名、样品描述、样品特性等。还有测序信息,例如测序平台、测序类型、 稳固大小等,其他的还包括发表文章的信息等,总之需要填很多的信息。 接下来还有一件重要的工作,就是要注册 ncbi 的账号,买 acbi, 获得账号和密码, 买这边用于管理上传后的数据,如果后面数据有更新,还需要使用同样的账号进行维护,所以要保证好。这个账号注册比较简单,和其他网上账号注册的类似,这里面就不介绍了。 在获得了账号之后,登录账号,我们就可以开始进行数据的上传工作了。首先我们来介绍一下上传金主数据, 刚刚到 ncba 上传页面,里面包含很多接收数据的数据库,根据 上传的数据类型进行选择。这里我们要上传金主文件,首先点击包围 pro, 再给他创建包围 progel 号和包围三拨号, 对某一个物种进行了金属车序,就是申请包围 pro 和包围上方号各一个,点击妙三文门诊。接下来就是需要填写各种信息, 经过几个步骤之后,最后点击萨博梅特。包一 pro 债务的创建成功,我们会得到一个包一 pro 债务的 id, 以 prg n a 字母为前缀,这个号码在后面会用到。第二步要做的就是获取 sbt 文件, 在汤普勒的报复页面填写一些信息,包括刚才得到的,包括在和 id 提交完了,就会生成一个以点 sbp 结尾的文件,汤普莱特点 svp, 将这个文件下载下来,接下来一个非常重要的工作就是使用 tbl two asn 产生 sqn 文件 还是科文文件,是上面还是 bp 文件,金主序列文件以及金主注视信息文件, tbl 的一个综合体,所以需要对这三个文件进行格式转换, 这个过程比较麻烦,容易出错。其中金主注视信息并不是必须的,也可以只上传金主序列还是 bt 文件,我们已经有了, 就是从 ncba 下载下来的化身文件,就是金主的拼接结果,后缀是点 f a, 点化肥或者点 f ic 等。 注意这里面有格式要求,金主文件序列中不能有 get, 每个文件不能超过一万条。 八十 a 序列,第一行是大于号开头,后面是描述信息,下面是序列信息,每一条序列长度不能超过八十个。制服 后车为 ppl 表格格式的金属注册文件,有固定的格式要求,这个文件比较麻烦, 此文件有五列,每列用泰国分割称为飞车。泰国这个文件是最为麻烦 一部。该文件必须包含编码基因的结构注视信息、非编码基因的结构注视信息和基因的功能注视信息等。 比如前面我们系列分析中的基因预测跟 cr 分析和基因功能出事,一旦做不好, ncbi 的工作人员就会发以迈偶反馈修改意见。对于飞车推广格式,需要注意以下几点。 首先,对每条序列的所有出任之前有一行额外的内容,例如 aj 十 w 的一 该行内容,后面所有出事都属于史高否一,一定不能遗漏飞车这个单词。飞车和史高否一 用空格来分割。每个飞车使用五行内容进行阐述,并分成两个部分。第一部分是飞车在序列上的结构信息,有三列,分别是该飞车的起始微点、结束微点和飞车名。而飞车在正义链上 折起指为点,以小于号结束为点。落在复印链上折起指为点,以大于号为结束为点, 让飞车为断裂的 cds 或者海审等信息。这有多行数据,但仅在其首行的第三列显示飞车的名字。 第二部分是飞车的功能处置信息, 使用第四到第五列 前面有三个推波键,第四列对应飞车的框里菲尔,第五列是匡尔菲尔的值,库尔菲尔就是对飞车的描述标签,如果有多个框里菲尔极其值,则用多行进行表述。 黑色和快乐肥标签的具体名称可以参考这个说明。 常用的飞车名有知音 mra、 cds、 f 三、五撇、 utr、 三撇、 utr、 tra、 rincra 等。 其中 a 三 r a 是指除了 t, r a 和 r a 以外的其余 c r a 基因的高尔夫标签,一般是基因。 第五列使用金主系统化的金 id, mr 和 cds 的 coutuber 标签一般使用 pro。 第五列是安儿注视的结果。 f 三的 colower 一般使用 mate, 例如一二三的有 tr 的框里边标签使用 nott。 第五列是相应的 rv 种类。 ncra 的括列篇标签必须有 ncra plus。 第五列则是 ncra 的类别,比如 mara, sra, scra 等,主要可以使用 nott 作为框里边的标签,即使可以随意标示 m, r a 和 cds 的朋友大额取持使用安儿注视的最终结果。 这三个准备 好了之后,就可以运行 ppl two as 生成 sk 文文件了。运行程序结束之后,会生成以序列 id 命名的三个文件,分别以点 sk 文、点 v a, l 和点 love 结尾。 其中点 sk 文件就是我们最后需要的文件。点 vl 文件可以查看转换过程是否出现问题,而点 logo 文件则用来监视转换的过程。一般来说,点 vl 文件大小为零,这转换过程没有问题,否则需要根据 报出信息进行修改。另外我们还需要一个 aj p 文件,这个文件主要是列出每条 catel 的信息,包括起始和中指位置。 app 文件只需要 要写一个简单程序就可以统计,并不是特别难。那么经过以上步骤之后,现在我们就可以使用频道 macose 的上传点 sk 文件,登录频道 marpods 网页 最下方输入框中填写信息,然后上传点 sk 文和 acp 文件。如果有符合 acbi 注视标准的注视文件也可以一起上传,不过生成满足 acbi 格式要求的文件非常麻烦。 接下来再填写上传的信息,最后点击沙巴梅特数据就上传完成了。 这一数据上传到 三 a 数据库比金主要容易一些,大家可以查看相应的说明文档即可。本次课程只是做一个简单的介绍, ncbi 也会不断修改上传数据的方式,具体执行过程中也可能与本课程介绍的稍有不同。 建议大家在上传数据之前请仔细阅读说明文档。
131基因学苑 11:14查看AI文稿AI文稿
11:14查看AI文稿AI文稿今天我们分享一下利用 ncbi 数据库下载芝麻的基因组。我们为什么要用 ncbi 去下载芝麻的基因组的话,首先就是因为我们在常见的植物基因组的网站上, 比方说 zambo, plant, 还有这个菲特字母上都没有找到芝麻的极左,因此呢我们在 ncbi 上找到了他。首先的话,首先我们需要打开 ncbi, 这有我收藏好的网站, 这样选择金,然后输入芝麻, 他这提示说这个金主的一个页面更新了,我们可以去新的页面去看一下。 搜索芝麻之后,他出现了四个版本,其中第一个版本的话他这标注了参考,而且的话,呃第一个的这个他这个水平呢已经到了染色体的水平,因此呢我们就啊点击第一个去看就行了, 这样的话就会有这个呃金主版本的一个 介绍,那最后面还有他这个染色体上的一个准备情况。 右侧的话还有关于这个金组的一个文章也可以去下载。接下来我们去下载这个金组数据, 我们只需要下载这个金主序列和这个注释的文件就可以了啊,当然呢我们也可以,嗯,下载一下这个 cds 序列和这个氨基酸序列,也就是蛋白质序列, 我们可以呃,到时候我们依据这个基因组序列和注视文件,呃,自己利用这个 tppos 软件进行这个 cds 的提取,然后呢也可以和这个下载的这个 cds 去进行一个比较, 点击下载就可以了, 已经下载完成了,然后我们打开就可以了 减压, 我们可以看到他提供的这个金组序列,他是按照染色体分开的,所以说我们首先呢需要把 这个金属训练的进行位置一下啊,就是把这个十几条染色体的训练进行一下合并,我们打开 tptos 软件, 点击 factor, tors 是里面有一个墨纸, 这些呢都是一个他的染色体训练。然后最后面的话就是有一个, 我们看一下这这个是什么文本,什么文件,然后呢这个文件的话就是说是有一些基因呢,它是没有注释到染色剂水平的,就是一些 staff 的片段, 我们也可以把这个去进行都合并到一起,他们都是精读训练, 我们设置一个输出的, 我们设置一个输出的文件, 点击开始, 哎已经合并完成了。 然后呢我们就进行这个 cds 的提取,首先我们需要输入 基因组织注视文件,点击出石化, 点击,在这我们可以看一下, 在这我们可以看一下如果我们汲取 cds 的话,他后面对应的就是一个 idid 是什么,因此呢我们就可以选择这个 id 作为这个标签,这样选择 cds 金组文件输入我们刚才木质完成的文件,设置输出文件 已经提取完成。 然后呢我们可以看一下这个 cds 的一个情况,一共有多少条基因啥的, 一共是三万五千多条基因。然后呢我们也可以可以拿我们提取的和这个下载的去进行一下对比,而这个就是下载的 cds 训练, 我们发现是没有任何差异的,我们直接下载的这个 cds 的训练,他也是一共有三万多条, 由于我们下载的这个我们提取的这个 cdsa 的这个 id 处有一个正负链的标记,我们可以对这个 id 进行一下简化, id 简化之后就是他这大于号后面就没有一个正负面的标记了, 然后其他的信息是不会改变的。接下来我们可以将批量的将这个 cds 序列翻译成蛋白质序列, 已经翻译完成, 我们发现蛋白质训练的这个数目的也是三万多条, 通过我们自己提取这个金组的 cds 训练,呃,然后呢再翻译成蛋白质训练去进行后续的分析, 这样子呢可以保证就是我们的京 id 来源于这个注视文件,后续的分析的话不会出现什么问题,因此呢后续的分析呢就是基于我们自己提取的这个 cds 训练和这个蛋白质训练去进行的。今天的分享呢就到这。
112樊二勤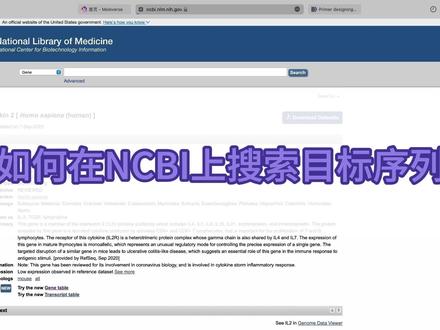 00:59查看AI文稿AI文稿
00:59查看AI文稿AI文稿首先我们可以在 ncba 上搜寻目的序列,在左边数据库中选择进, 我们以白细胞激素二者基因为例,在输入框中搜索 i l。 二,点击搜索,右边有一个筛选物种的选项卡,今天我们筛选出人员的 i l 二基因。 好,我们点击 l 二者选项卡进入,然后下拉找寻到 m, i, a 和蛋白相关的信息, 这里显示是 m, i, a 和蛋白的一些信息。点击这个 n n m 开头的编号进入,可以查看 m n, a 对应的 c, d, n, a 序列的一些详细信息。 这里我们可以点击 c, d, s 序列,又显示出了整个 m i 里面 c, d, s 的区域,我们选中复制。
129术也VECVERSE 03:25查看AI文稿AI文稿
03:25查看AI文稿AI文稿不要放过莫德纳,一个法人类的美国疫苗公司。三月十七日,英国 delex boss 点 uk 揭露美国莫德纳合成了新冠病毒的文字完全不是空穴来风, 作者给出了合理怀疑莫德纳制造新冠病毒的线索。如果你还不相信新冠病毒是人类干预下生产的病毒,下面步骤告诉你如何根据基因组对比验证作者的合理推断。 第一步,到美国国家售技术信息中西网站连 cba 查找基因组。第一个基因组及时新冠病毒的完整基因遗传密码 a c b i, 网站代码是 in c 零四五五一二点二,这是在自然杂志发布,根据二零一九年十二月的新冠病毒样本的中国科学家发现并公布的完整的基因组序列。第二个基因组在大自然存在了上一年的蝙蝠冠状病,读瑞特一三的基因组编号 mn 九九六五 三二,日期为二零零三年七月二十四日原始蝙蝠病毒。第二步,基因对比用美国国家生物技术信息中心网站提供的不赖色工具对两组金库做完整对比分析,可以发现,两个基因组完整的遗传密码百分之九十六相互匹配,只是存在下面的基因片段的差异。 基因组的不匹配情况如下,仅有一个字母不匹配的有九百九十五处,连续有两个字母不匹配的有二十四处。连续有三个字母不匹配的有四处,连续有十二个字母不匹配的仅有一处。此处位于基因组两万三千五百七十六行,存在连续十二个字母的不匹配。 正是这十二个连续的不匹配基因泄露了天机自然突变,通常一次发生一个字母。因此,要确定新冠病毒是否是人造的,我们只需要查看十二个字母的排列。这对 对于在两万九千八百七十七个字母的基因组中恰好相邻发生的一系列随即突变来说,几率太小了。而且这发生在 rat 一三被测序的二零一三年和新冠病毒出现的二零一九年之间的六年时间内这么短的时间。 所以这十二个字母,也就是十二个剪辑是从另一个基因组中插入的,人为插入的可能性几乎是百分之一百。 第三步,核对专利库同样去美国国家生物技术信息中心的专利库进行查询。针对与十二个基因序列相关的专列,经过查询可以发现,在引用插入的所有专利中,只有莫得那专利与新冠病毒中的相关的整个十九个核苷酸序列匹配。 在新冠病毒到来之前提交的其他专利并没有没有完整的十九个字母序列。莫德纳是唯一一家在二零一九年之前提交人类使用基 插入物的公司。他们在二零一三年十二月十六日至二零一六年二月四日期间使用基因插入物申请了五项专利,这也是他们在二零一九年十月新冠疫情爆发之前提交了最后五项专利。 在英国 delex boss 点 uk 的揭露文章中,媒体作者提供了完整的可核对的证据链,指正莫德纳用插入基因制造了新冠病毒。 如果有企图为莫德纳喜庆嫌疑的公知或者专家们,请拿出不是莫德纳制造病毒的科学证据来反驳上面的揭露,文章应该以科学的态度去寻根溯源,而不是立场先行栽赃嫁祸。 对于英国 delex boss 点 uk 揭露莫德纳制造新冠病毒的信息,您怎么看?欢迎评论区留言。
1520普吉龙哥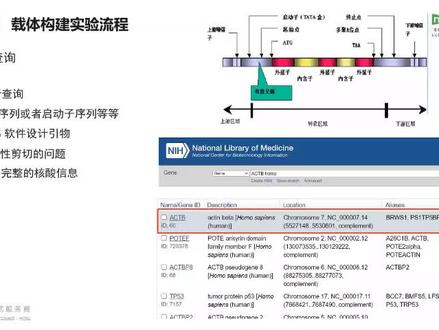 01:55查看AI文稿AI文稿
01:55查看AI文稿AI文稿那么首先就是我们的这个训练查询啊,那么还在训练查询的时候,我们还要做一个事情,就是你要有空仔,那么有些实验室老师坑没有,那么你可以去啊,给自己去买一些空仔,或者是实验室常用的一些空仔啊,都 ok 的。 那么我们这这边只能训练查询,就是说我们自己要构建的这样一个目的基因的这样的一个查询啊,那么是在 ncpi 上面去进行查询的,那么现在呃构建的比较多的像一些啊各表达的一些载体, 那么我们可能需要找一下他的这个呃 cids 序列,那么或者是一些启动子分析载体,那么需要找到小一下他的这种启动子序列的,对吧?我们都是需要在 ncba 里面去进行查找的 啊,因为他你查找出来之后,这样才便于我们后面去设计引物啊,并且对于一些经表达展点来 说,你的这个呃有一些基因它是存在的这种多种的可比性剪切的啊,那么呃我们可能需要需要看一下,如果你想要主要的这样一个呃可比性剪切的话和其他的话,你的这个营物设计其实还是 呃还是需要呃注意一下的好,那么我们就是还有就是用传我的这个软件啊,直接去进行设计引用,或者是用那个四代部记啊,都可以的啊,只要是能够让你看训练然后设计就 ok 了啊。咱们这边有刚刚有收到注意可微信解切的一些问题, 那么甚至甚至有的基因可能有上十个,对吧?他会表达不同的这样一个转录本,那么这个是需要我们注意的,嗯,我们可以在接半个里面可以得到一些完整完,比较完整的啊,一些这种核酸的这样一个信息啊。那么这个也便于我们在 训练查询过后,后面做一些扩增的时候,那么我们也知道我们要的目的金它的片段的片段大小大概是多少,对吧?因为有的可能会出现你扩增到啊金足上面的一些问题,那么这也是我们前期呃,查询是一定要去做的。
63爱必信生物 10:46查看AI文稿AI文稿
10:46查看AI文稿AI文稿去年时候给大家录制的一个操作视频,那么在评论区里面呢,看到了有很多的 很多的问题,我们这里有没有什么方法可以把检索到的序列号的来源地也快速批量展示出来呢?然后是博主使用的是什么软件,打开查卖格式的, 那么后面的还有大佬导出的文件只能固定几个列吗?我想把学名加群怎么弄,其实有好多都在问我怎么办,然后呢 然后呢给大家展示展示一下整体的石油 情况,一个是套盆,然后里面呢还有是吹磨案子,这是三个一代测去之后呢可以 根据呢质量测试结果的质量呢进行一个自动的两端的去除,那么这里可以选择。然后这几个功能呢就不给大家介绍了,还有几个几枪功能,我也给他指挥了这些技能呢,这些 这些那个模块呢,大家可能用不到,那么今天呢就是我把之前做的一些工作呢,通过这个软件呢 让他更快的去实现,那么就给大家开始演示一下,首先呢这有批量的局不拉丝,这九条序列,这九条九个物种吧,可以这么认为。 然后在 ncba 上官网上进行粘贴,粘贴完了之后呢点不拉死他,其实这一步呢也可以用程序实现,但是呢 ncbi 上官网呢有一个说明,就是如果一个程序呢在 一天之内呢提交有一百次的话,那么他会把这个 ip 呢给现放到慢的那个资源序列里面弄,所以说我们还是不要还是通过外部网页来进行,这样会 安全一些。等比对出来之后呢,我们选择这个螳螂的哦,选择黑的 table csv, 这个在之前的视频当中呢,也给大家展示过, 从下载时下载完了之后呢,我看啊,我们该用, 嗯,可以打开看一看, 相当于一号,一号的物种呢,他比对到了有九十八十九个,那么这对应他的,这是按照思考的排序,嗯, 然后呢,我们可以去他这是匹配到的他的对应的这个检索号,核酸的检索,我这核酸系列核酸检测号,这是二号物种,以及一共是九个,那么在一千一百六十三个里面去 寻找出来我们那个需要的,我一般都是按照 score 排序, 先不保存,然后好好打开一下,打开之后呢,看一下这个解释一下啊。 top nanabetchantress, 其实,呃, 拿到他对应的解锁号之后呢,我们去获取淘汰他的内容,那么先看一看啊,先把这个先捡走,你可以在这个工作目中进行操作,嫌他乱,所以说放到临时文件里,这有一个线索哎, 他可以把他直接拖进来,然后呢一个白色扣一个白按得体,这可以选这个呢,我是勾选的欧内这个核酸裤啊,这个根据登录后面直直接检索这个物种的信息, 他也可以用蛋白裤,还有很多,我这里呢参数呢,我就是设定的是核酸裤,我限制死了,因为 啊不想增加那么多工作量啊,其实就改个参数就行了,如果大家有需要的话,我可以把这个功能这块给大家放开,然后呢套喷一望着你,我们来做几次,我想要套喷一点开始,那么他就会 进行一次通过网络上的传输,我们来看一下,哎,已经拿到了,首先呢他会自动下载一个,呃,这是 top 一里边的这五种信息,这样是 这个插麦耳文件,所以说有的同学呢就是问我这个插麦耳的文件怎么打开,所以说我就在这里的软件呢,是自动给生成了一个呃,一一对应好的最终结果啊。电视看,这是 一号,他对应的嗯,登录号,然后这个登录号,这是最开始的 csv 那个,我们那个布拉斯出来这些 是告知了,然后有多少 gap, 是从从多少到多少匹配多少,这个是那个五种的 top c id, 那么我们看一下他是什么,他是小麦,这是小麦,然后包括呃下面的对应的每个都是什么,以及他的描述,他的定义, 然后他后面的是跟着就是比对好的这个序列,这是这个来自于这个物种的硬气。边上公布的这些序列已经就放到这里了, 这是套个一,然后我们看一下,嗯,当然了你也可以看一下,套个二,然后点,同样套个三,再点,然后套 四,接着点啊,这是走了一个多县城啊。然后就是,嗯,如果这个完成的话,他这个 显示状态显示为四个 c 的,四个 c 的,然后或者是如果有一个文件下载,如果特别大的话,比如说你按照九个按照速高之,然后点开始的话,他会特别慢,而且下载这个也出不来,之后这个出来里面的序列特别长,而导致这个 程序这块的阻塞,所以说你可以点看守,我就不要走这块了。这个是仅仅做了一些简单的一个测试啊, 我们去看一下,按照这个排序查看,按照详细列表,按照日期,日期道序,这个是出来的,我们看 每个文件都都用在对应的他的拆卖文件,可以大家去做个脚印,这是四个,看到没,这 top 四, top 四是对应的是这几个,那么后面的信息 这是一个,呃,这个软件里面呢,还有我开发这个小工具里面还有一个盖子赛克白 id, 这个呢,这个用法呢,大家可以去看以前的视频,我也做了说明,然后 combas 有个范儿,这个是测序文件的,批量的, 批量的组合起来,然后方便使用。在这里 ncba 上呢,也就说这个格式整理,我在这里用了他 combasecophoney, 用他来做了一个整理,可以拼起来这么多不同格式的文件放到这里,然后进行 ncba 的批量,不拉丝的。 然后再讲一下这个吹木案子,这个测序在测序里面呢,我有收到这些测序文件呢。啊,吹木案子,他的 ab 一格是文件, ab 一格是, 嗯,就是他的测序风图,这里呢就是我设计的几个值,那么十呢?如果选择十的话呢,这个概率呢?有百分之九十的可能性是测序的准确度,那么三十以上就百分之九十九点九九九,后面四个九,那么 这也是我查一些资料知道的,那么五盒把我自己设定的,嗯,所以说在这里的话,就是目前是设计的,是大家去点选,嗯,本来想做成输入数字的话,那么我正好前面的有这个模板,我就直接套 好用了。所以说五和八,比如说我们选择一个五,然后希莱克特选择地盘 tamp 测序,就这个文件夹里面,我要把批量的 ab 这个三格测取的文件呢,全部给他自动来切除的话,那么我们会质量高一点的。如果都大于,这是大于等于三十用,如果大于等于五的话,点凹凸,然后已经结束了。点确定 啊,软件还设置自动跳转啊。这个是为了方便大家去检索一下,他就是生成的文件是在当前的目录下 啊,这是生成完的,我们按照名称排下去,就是一一对应。好的,嗯,我处理好的,把这些中括号什么都处理的是方便。 嗯,一会给大家再展示一下啊。展示一下,这是测序文件,我们测序拿到这个之后呢,我先比对一下, 嗯,就这样,刚才我做过测试看一下,那么去除了前面的有十一个, 那后面呢,会去说多一些,呃,九百一以后,这是这个值大于五的,那么大于二十的话呢,当然去说会更多。 然后这里的话再给你展示一下 combaocombaoxicophil, 比如说我已经吹木案子了,吹木之后呢,我可以再选择这边地盘 tempu 测去,就这个文件夹里面没选。这是赛克,这是公司返回来之后,他自己有一个 c 款式,他从 其实就从 ab 的转直接提出来序列文件,那么这里呢,我用 txt, 就是我刚才自己去去母完了之后呢这个,然后我啃白闹,然后他最后会合成到一个法塞格式文件,这也做了一个弹窗, 这个发 c 大家看出来没,这是他的名字,然后后面对应的他的序列,这样的话,你可以拿着这个 ctrla 复制完了之后呢,在这里再粘贴,这里就直接装不拉丝就完事了 啊,整个整个这个软件呢?这样操作下来是非常快的,如果大家还是有什么不懂的话,可以在评论区里面问我吧!
12河农博士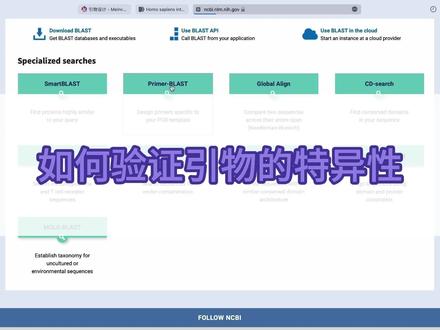 01:29查看AI文稿AI文稿
01:29查看AI文稿AI文稿接下来我们需要验证引物的特异性,我用 n c b i 的 primer blast 网站给大家演示。首先打开 blast 往下翻,找到 primer blast 这个网页可以帮助我们验证引物的特异信。 然后将我们的引物分别粘贴着入 primar parameters 这里的输入框,正向引物输入到第一行, 反向引物输入到第二行。这里的顺序先不要搞错,然后再给这一块选项卡。这里这里的物种请选择我国募集基因的来源物种,这里默认是人。 然后我选的也是人员的白细胞激素二基因,所以不用改,点击 get promise two thousand years late 最好选取的是具有唯一的 p c r 扩增结果的引物队,并且这个 p c r 结果啊是我们的目的基因。 然后可以看到我们选取的这对引物的特异性还是非常不错的,只有一个扩增结果,并且这个扩增结果也确实是我们的目的基因就是人员的白细胞激素二。所以我们就可以将这对引物交给金公司合成。接下来就可以尝试进行 qpcr 实验了。
138术也VECVERSE





猜你喜欢
最新视频
- 1.2万乐乐